DNA Sequence Classification based on Milvus
Author: Mengjia Gu, a data engineer at Zilliz, graduated from McGill University with a Master degree in Information Studies. Her interests include AI applications and similarity search with vector databases. As a community member of open-source project Milvus, she has provided and improved various solutions, like recommendation system and DNA sequence classification model. She enjoys challenges and never gives up!
DNA sequence is a popular concept in both academic research and practical applications, such as gene traceability, species identification, and disease diagnosis. Whereas all industries starve for a more intelligent and efficient research method, artificial intelligence has attracted much attention especially from biological and medical domain. More and more scientists and researchers are contributing to machine learning and deep learning in bioinformatics. To make experimental results more convincing, one common option is increasing sample size. The collaboration with big data in genomics as well brings more possibilities of use case in reality. However, the traditional sequence alignment has limitations, which make it unsuitable for large data. In order to make less trade-off in reality, vectorization is a good choice for a large dataset of DNA sequences.
The open source vector database Milvus is friendly for massive data. It is able to store vectors of nucleic acid sequences and perform high-efficiency retrieval. It can also help reduce the cost of production or research. The DNA sequence classification system based on Milvus only takes milliseconds to do gene classification. Moreover, it shows a higher accuracy than other common classifiers in machine learning.
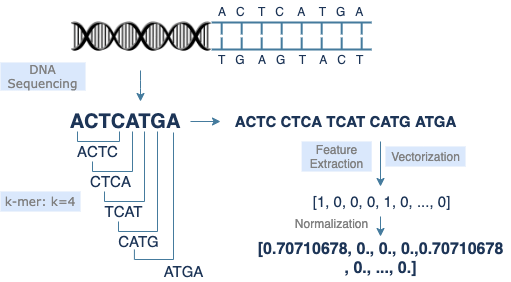
A gene that encodes genetic information is made up of a small section of DNA sequences, which consists of 4 nucleotide bases [A, C, G, T]. There are about 30,000 genes in human genome, nearly 3 billion DNA base pairs, and each base pair has 2 corresponding bases. To support diverse uses, DNA sequences can be classified into various categories. In order to reduce the cost and make easier use of data of long DNA sequnces, k-mer is introduced to data preprocessing. Meanwhile, it makes DNA sequence data more similar to plain text. Furthermore, vectorized data can speed up calculation in data analysis or machine learning.
 1.png
1.png
k-mer
The k-mer method is commonly used in DNA sequence preprocessing. It extracts a small section of length k starting from each base of the original sequence, thereby converting a long sequence of length s to (s-k+1) short sequences of length k. Adjusting the value of k will improve the model performance. Lists of short sequences are easier for data reading, feature extraction, and vectorization.
Vectorization
DNA sequences are vectorized in the form of text. A sequence transformed by k-mer becomes a list of short sequences, which looks like a list of individual words in a sentence. Therefore, most natural language processing models should work for DNA sequence data as well. Similar methodologies can be applied to model training, feature extraction, and encoding. Since each model has its own advantages and drawbacks, the selection of models depends on the feature of data and the purpose of research. For example, CountVectorizer, a bag-of-words model, implements feature extraction through straightforward tokenization. It sets no limit on data length, but the result returned is less obvious in terms of similarity comparison.
Milvus can easily manage unstructured data and recall most similar results among trillions of vectors within an average delay of milliseconds. Its similarity search is based on Approximate Nearest Neighbor (ANN) search algorithm. These highlights make Milvus a great option to manage vectors of DNA sequences, hence promote the development and applications of bioinformatics.
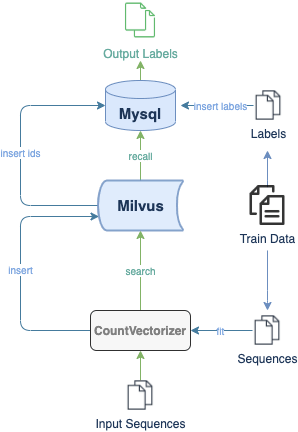
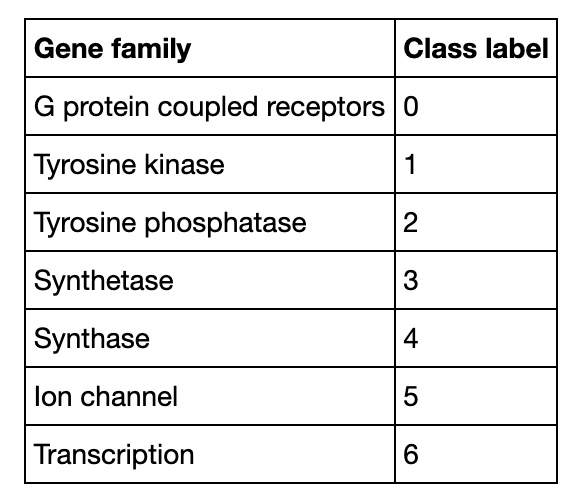
Here is a demo showing how to build a DNA sequence classification system with Milvus. The experimental dataset includes 3 organisms and 7 gene families. All data are converted to lists of short sequences by k-mers. With a pre-trained CountVectorizer model, the system then encodes sequence data into vectors. The flow chart below depicts the system structure and the processes of inserting and searching.
 1.png
1.png
Try out this demo at Milvus bootcamp.
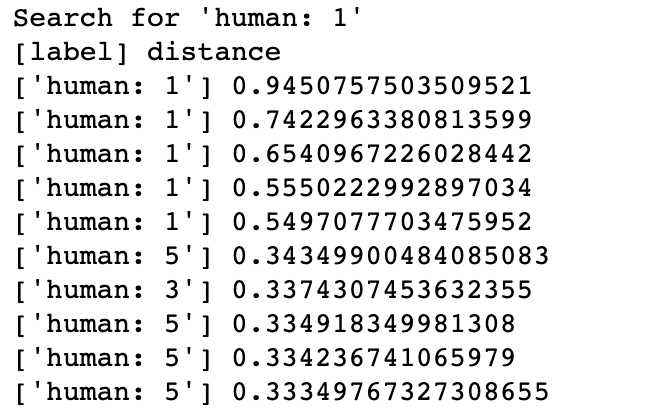
In Milvus, the system creates collection and inserts corresponding vectors of DNA sequences into the collection (or partition if enabled). When receiving a query request, Milvus will return distances between the vector of input DNA sequence and most similar results in database. The class of input sequence and similarity between DNA sequences can be determined by vector distances in results.
# Insert vectors to Milvus collection (partition "human")
DNA_human = collection.insert([human_ids, human_vectors], partition_name='human')
# Search topK results (in partition "human") for test vectors
res = collection.search(test_vectors, "vector_field", search_params, limit=topK, partition_names=['human'])
for results in res:
res_ids = results.ids # primary keys of topK results
res_distances = results.distances # distances between topK results & search input
DNA Sequence Classification Searching for most similar DNA sequences in Milvus could imply the gene family of an unknown sample, thus learn about its possible functionality. If a sequence is classified as GPCRs, then it probably has influence in body functions. In this demo, Milvus has successfully enabled the system to identify the gene families of the human DNA sequences searched with.
 3.png
3.png
 4.png
4.png
Genetic Similarity
Average DNA sequence similarity between organisms illustrates how close between their genomes. The demo searches in human data for most similar DNA sequences as that of chimpanzee and dog respectively. Then it calculates and compares average inner product distances (0.97 for chimpanzee and 0.70 for dog), which proves that chimpanzee shares more similar genes with human than dog shares. With more complex data and system design, Milvus is able to support genetic research even on a higher level.
search_params = {"metric_type": "IP", "params": {"nprobe": 20}}
Performance
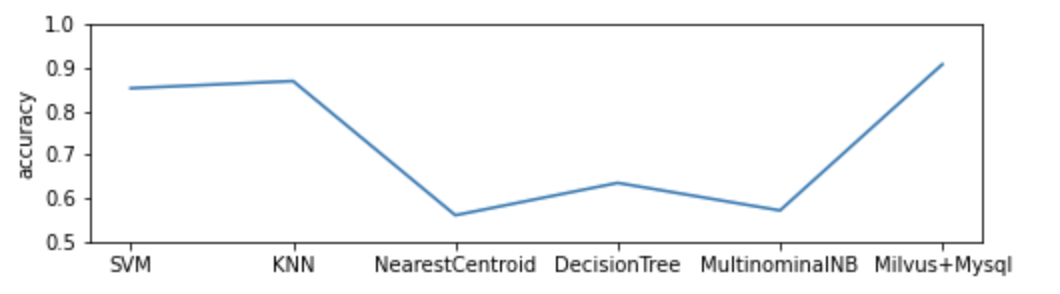
The demo trains the classification model with 80% human sample data (3629 in total) and uses the remaining as test data. It compares performance of the DNA sequence classification model which uses Milvus with the one powered by Mysql and 5 popular machine learning classifiers. The model based on Milvus outperforms its counterparts in accuracy.
from sklearn.model_selection import train_test_split
X, y = human_sequence_kmers, human_labels
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
 1.png
1.png
With the development of big data technology, vectorization of DNA sequence will play a more important role in genetic research and practice. Combined with professional knowledge in bioinformatics, related studies can further benefit from the involvement of DNA sequence vectorization. Therefore, Milvus can present better results in practice. According to different scenarios and user needs, Milvus-powered similarity search and distance calculation show great potential and many possibilities.
- Study unknown sequences: According to some researchers, vectorization can compress DNA sequence data. At the same time, it requires less effort to study structure, function, and evolution of unknown DNA sequences. Milvus can store and retrieve a huge number of DNA sequence vectors without losing accuracy.
- Adapt devices: Limited by traditional algorithms of sequence alignment, similarity search can barely benefit from device (CPU/GPU) improvement. Milvus, which supports both regular CPU computation and GPU acceleration, resolves this problem with approximate nearest neighbor algorithm.
- Detect virus & trace origins: Scientists have compared genome sequences and reported that COVID19 virus of probable bat origin belongs to SARS-COV. Based on this conclusion, researchers can expand sample size for more evidence and patterns.
- Diagnose diseases: Clinically, doctors could compare DNA sequences between patients and healthy group to identify variant genes that cause diseases. It is possible to extract features and encode these data using proper algorithms. Milvus is able to return distances between vectors, which can be related to disease data. In addition to assisting diagnosis of disease, this application can also help to inspire the study of targeted therapy.
Milvus is a powerful tool capable of powering a vast array of artificial intelligence and vector similarity search applications. To learn more about the project, check out the following resources:
Try Managed Milvus for Free
Zilliz Cloud is hassle-free, powered by Milvus and 10x faster.
Get StartedLike the article? Spread the word



